
- 特色分析
- 基因组分析
- 转录组分析
- 基因图谱
- AI育种模型构建
- 近期发表文章
特色分析 – 主要针对医院及高校的科研团队,从实验设计、样本收集、测序标准分析、个性化分析到结果解读等科研过程提供全方位或是某一过程的服务。
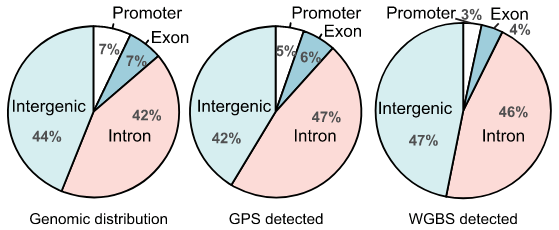
甲基化测序分析
基于全基因组水平,实现单碱基分辨率的甲基化位点定位。进行差异甲基化分析、相关基因GO/KEGG分析、PCA多样本甲基化变化规律。
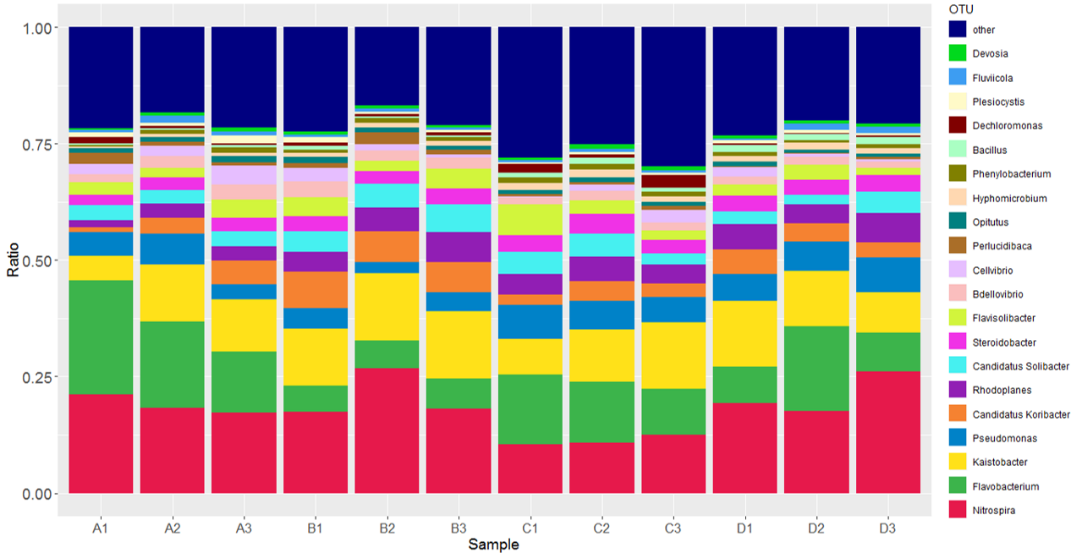
16S rDNA测序分析
对群落的16S rDNA可变区序列进行高通量测序。进行物种注释、物种组成分析、物种差异分析、环境因子关联分析、进化分析以及菌群的功能预测等。
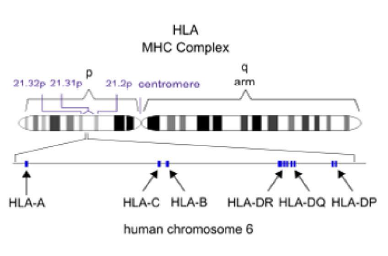
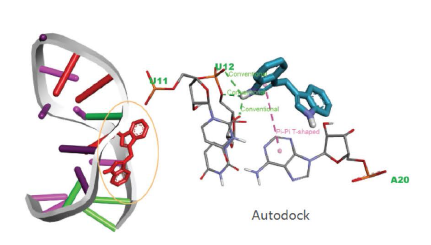
HLA分型
对HLA区域进行捕获测序。进行从头组装HLA 基因,HLA型别分析、注释,基于统计学检验的显著性分析。
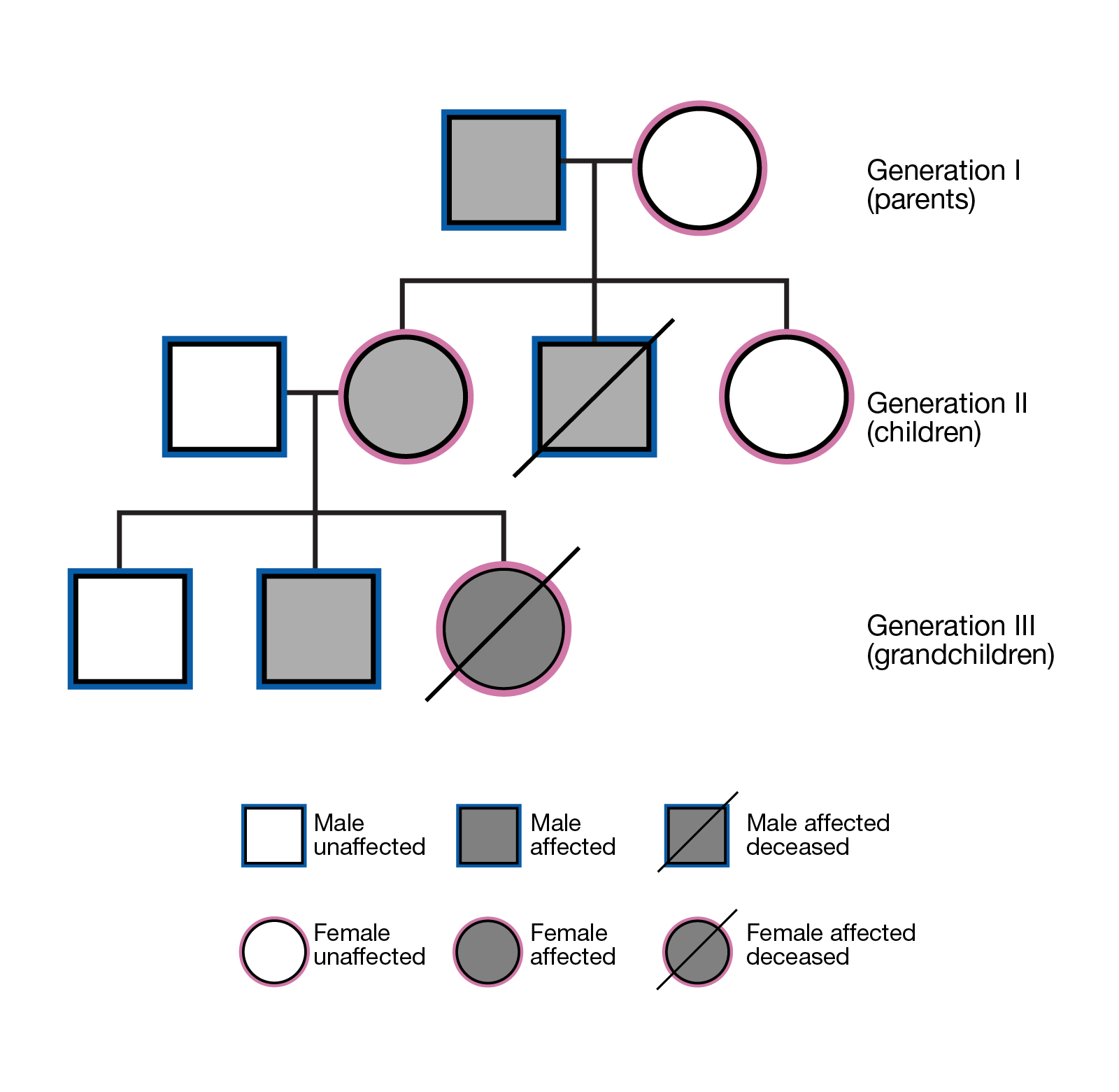
家系连锁分析
基于显性遗传模式和隐性遗传模式的遗传特点筛选与疾病相关的变异。基于不同世代的个体之间存在的基因传递关系,筛选与家族遗传病相关的变异。
基因组分析 – 根据测序结果,比对参考基因组,检测样本基因组的单核苷酸多样性变异,插入缺失变异,拷贝数变异及结构变异,并对无参考基因组的物种构建对应参
考基因组。
全基因组重测序分析
利用最新参考基因组对具有参考序列的不同个体进行全基因组测序。与参考序列进行比对、统计测序深度及覆盖度;进行SNP、InDel、CNV、SV的检测、注释。
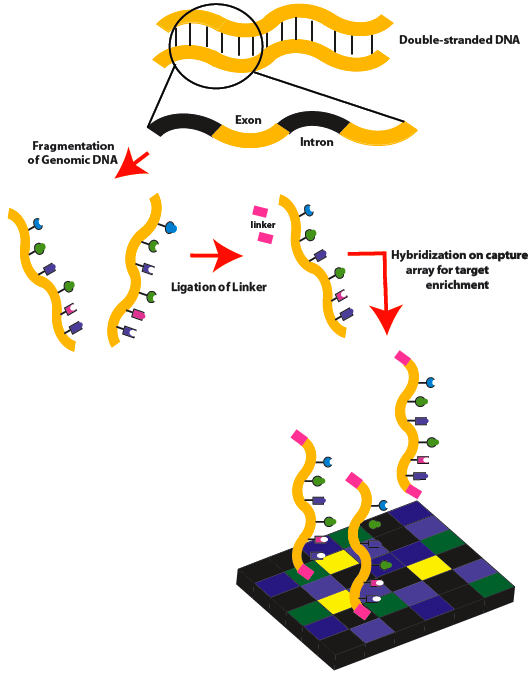
外显子组测序分析
利用序列捕获或者靶向技术将全基因组外显子区域DNA富集后再进行高通量测序。与参考序列进行比对、统计测序深度及覆盖度;进行SNP、InDel、CNV、SV的检测、注释。
简化基因组测序及QTL定位分析
利用限制性核酸内切酶打断基因组DNA,对待定片段进行高通量测序获得海量遗传多态性标签序列来充分代表目标物种全基因组信息。与参考序列进行比对、统计测序深度及覆盖度;进行SNP、InDel、CNV、SV的检测、注释;进行QTL定位分析
全基因组survey
基于小片段文库的低深度测序数据,通过K-mer分析,从而有效的评估基因组大小、GC含量、杂合度及重复序列含量等信息,全面了解某一物种基因组特征,为后续的全基因组Denovo测序组装策略的制定提供依据。
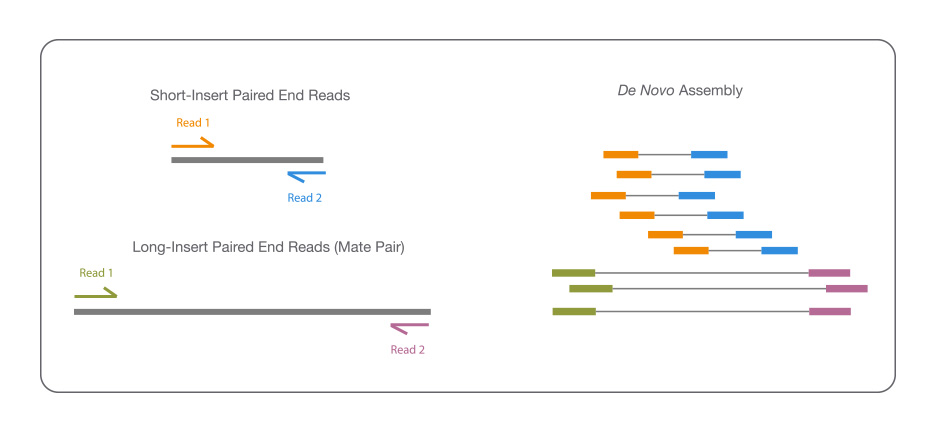
De novo组装
对基因组序列未知或没有近源物种基因组信息的某个物种,对其不同长度基因组DNA片段及其文库进行序列测定,然后进行拼接、组装和注释,从而获得该物种完整的基因组序列图谱。
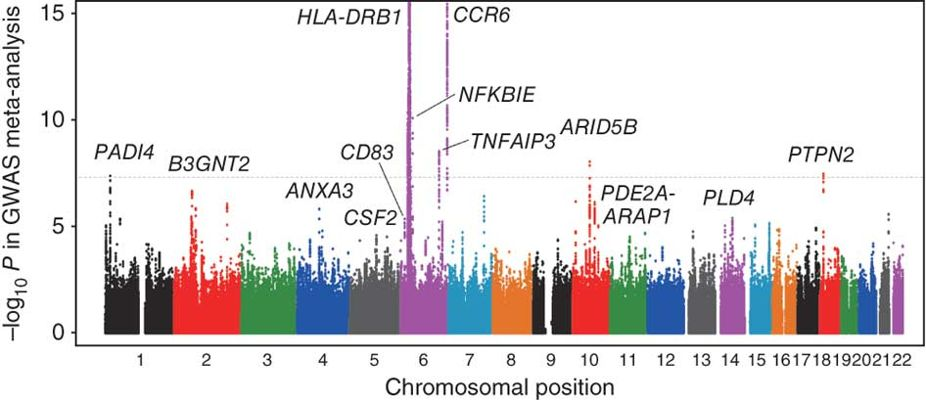
全基因组关联分析(GWAS)
通过高通量测序找到染色体上的变异位点,研究这些变异位点与疾病或其他性状的关联。全基因组关联分析是对具有丰富遗传多样性的群体的每个个体进行全基因组重测序,结合目标性状的表型数据,基于一定的统计方法进行全基因组关联分析,可以快速获得影响目标性状表型变异的染色体区段或基因位点。利用NGS数据提供的海量标记,全方位的解析性状与基因之间的关系,精确定位目标性状。
转录组分析 – 根据测序结果比对参考基因组,整理转录本及基因水平表达量,并预测新转录及基因融合结果。
![]()
转录组测序分析
对特定组织或细胞在某个特定状态下转录的所有mRNA进行测序。进行差异表达基因分析、GO分析和KEGG通路富集,分析得到的差异基因参与的功能或通路。
![]()
lncRNA测序分析
利用高通量测序技术进行lncRNA测序,进行lncRNA分析,分析其与特定生物学过程的关系。进行lncRNA拼接组装、lncRNA位点筛选、lncRNA编码潜能分析、lncRNA靶基因预测、lncRNA保守性分析及差异表达分析等。
![]()
Small RNA测序分析
对样本中的miRNA、siRNA、piRNA等进行高通量测序。进行small RNA分类注释、碱基编辑分析、差异表达分析、靶基因分析及差异sRNA靶基因GO、KEGG富集。
![]()
全转录组测序分析
对特定组织或细胞在特定状态下转录出的所有转录本的总和,包括mRNA和所有的non-coding RNA,通过构建small RNA文库和去rRNA的链特异性文库,分析mRNA和non-coding RNA 的表达和调控关系。
![]()
外泌体测序分析
对外泌体RNA进行测序,获得外泌体RNA的信息。进行差异表达分析、多组样本PCA分析、差异表达模式聚类、靶基因预测。
为珍贵动植物构建参考序列图谱,丰富大基因组参考序列数据库的数据。
基因组survey
根据二代测序结果,预测样本基因组的大小、杂和度、重度序列比例等信息。
contig构建
根据三代测序结果,将序列初步组装为contig形式,并利用二代测序结果校正contig。
scaffold构建
结合bionano结果,将contig组装为scaffold形式。
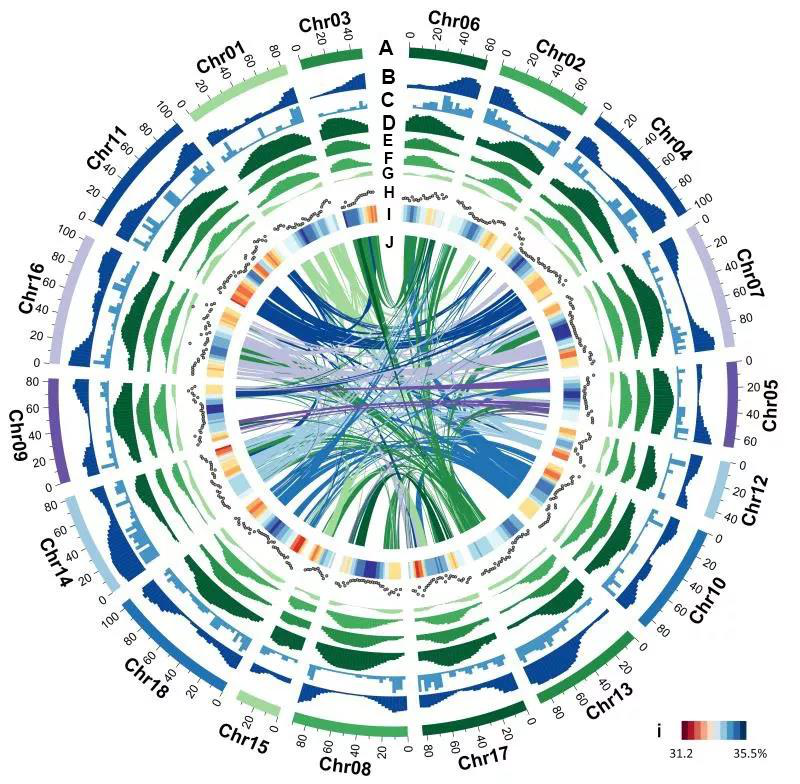
Hi-C协助组装
结合Hi-C测序结果,将scaffold组装成更完整的基因组。
基因组注释
结合样本亲缘关系较近物种基因组与样本转录组测序数据,注释样本基因组各区域功能。
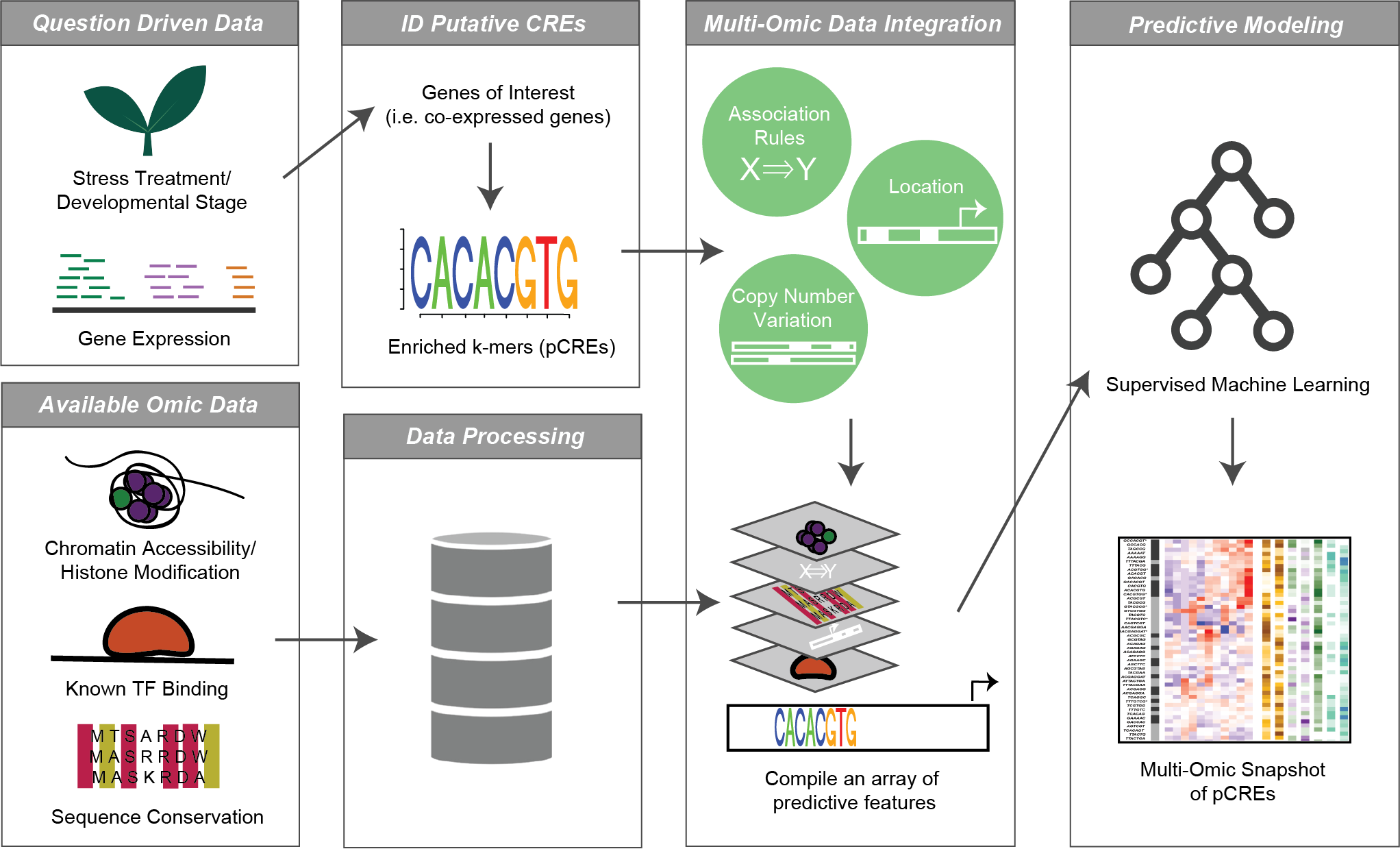
AI育种模型构建
在全基因组层面上建立机器学习预测模型,实现智能、高效、定向培育新品种。
案例分享
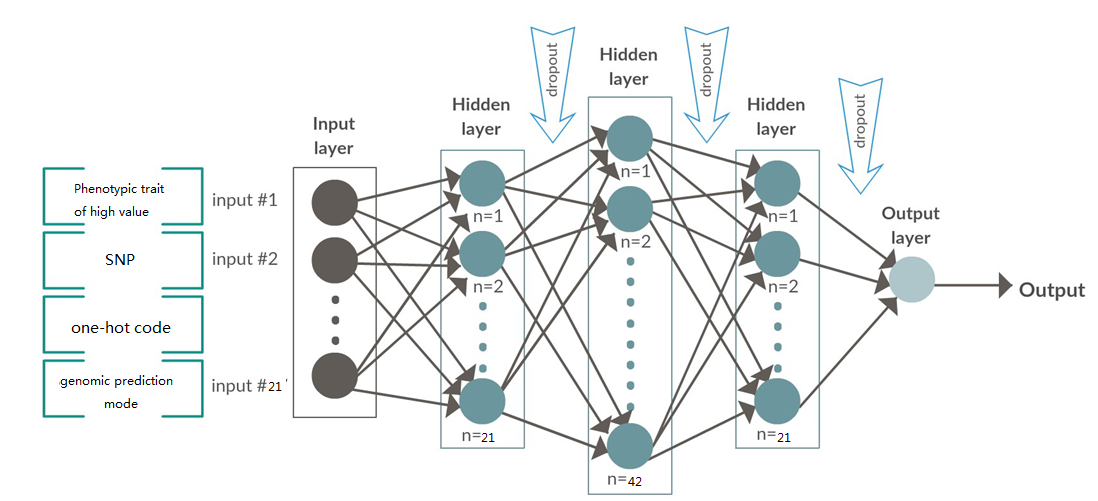
基于深度卷积神经网络的基因组选择
明领基因携手高校农业团队对企业大白猪的100kg日龄,100kg背膘厚和母猪乳头数3个性状为分析对象,结合猪50K基因芯片分型数据,以加性模型为基础,通过结合深度学习和BLUP形成的集成模型,开发一套半监督自主学习的深度卷积神经网络的算法,训练基因型-表型预测模型和基因型-表型模型,用在种猪分子育种生产系统中。从根本上缩短了育种时间和成本,同时预测准确性得到精确的提升。

Chromosome-level genome assembly of goose provides insight into the adaptation and growth of local goose breeds
Ackground:Anatidae contains numerous waterfowl species with great economic value, but the genetic diversity basis remains insufficiently investigated. Here, we report a chromosome-level genome assembly of Lion-head goose (Anser cygnoides), a native breed in South China, through the combination of PacBio, Bionano, and Hi-C technologies.
Findings:The assembly had a total genome size of 1.19 Gb, consisting of 1,859 contigs with an N50 length of 20.59 Mb, generating 40 pseudochromosomes, representing 97.27% of the assembled genome, and identifying 21,208 protein-coding genes. Comparative genomic analysis revealed that geese and ducks diverged approximately 28.42 million years ago, and geese have undergone massive gene family expansion and contraction. To identify genetic markers associated with body weight in different geese breeds, including Wuzong goose, Huangzong goose, Magang goose, and Lion-head goose, a genome-wide association study was performed, yielding an average of 1,520.6 Mb of raw data that detected 44,858 single-mucleotide polymorphisms (SNPs). Genome-wide association study showed that 6 SNPs were significantly associated with body weight and 25 were potentially associated. The significantly associated SNPs were annotated as LDLRAD4, GPR180, and OR, enriching in growth factor receptor regulation pathways.
Conclusions:We present the first chromosome-level assembly of the Lion-head goose genome, which will expand the genomic resources of the Anatidae family, providing a basis for adaptation and evolution. Candidate genes significantly associated with different goose breeds may serve to understand the underlying mechanisms of weight differences.
Molecular Phylogenesis and Spatiotemporal Spread of SARS-CoV-2 in Southeast Asia
Background: The ongoing coronavirus disease 2019 (COVID-19) pandemic has posed an unprecedented challenge to public health in Southeast Asia, a tropical region with limited resources. This study aimed to investigate the evolutionary dynamics and spatiotemporal patterns of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the region.
Materials and Methods: A total of 1491 complete SARS-CoV-2 genome sequences from 10 Southeast Asian countries were downloaded from the Global Initiative on Sharing Avian Influenza Data (GISAID) database on November 17, 2020. The evolutionary relationships were assessed using maximum likelihood (ML) and time-scaled Bayesian phylogenetic analyses, and the phylogenetic clustering was tested using principal component analysis (PCA). The spatial patterns of SARS-CoV-2 spread within Southeast Asia were inferred using the Bayesian stochastic search variable selection (BSSVS) model. The effective population size (Ne) trajectory was inferred using the Bayesian Skygrid model.
Results: Four major clades (including one potentially endemic) were identified based on the maximum clade credibility (MCC) tree. Similar clustering was yielded by PCA; the first three PCs explained 46.9% of the total genomic variations among the samples. The time to the most recent common ancestor (tMRCA) and the evolutionary rate of SARS-CoV-2 circulating in Southeast Asia were estimated to be November 28, 2019 (September 7, 2019 to January 4, 2020) and 1.446 × 10−3 (1.292 × 10−3 to 1.613 × 10−3) substitutions per site per year, respectively. Singapore and Thailand were the two most probable root positions, with posterior probabilities of 0.549 and 0.413, respectively. There were high-support transmission links (Bayes factors exceeding 1,000) in Singapore, Malaysia, and Indonesia; Malaysia involved the highest number (7) of inferred transmission links within the region. A twice-accelerated viral population expansion, followed by a temporary setback, was inferred during the early stages of the pandemic in Southeast Asia.
Conclusions: With available genomic data, we illustrate the phylogeography and phylodynamics of SARS-CoV-2 circulating in Southeast Asia. Continuous genomic surveillance and enhanced strategic collaboration should be listed as priorities to curb the pandemic, especially for regional communities dominated by developing countries.

H NMR and UHPLC/Q-Orbitrap-MS-Based Metabolomics Combined with 16S rRNA Gut Microbiota Analysis Revealed the Potential Regulation Mechanism of Nuciferine in Hyperuricemia Rats
Hyperuricemia seriously jeopardizes human health by increasing the risk of several diseases, such as gout and stroke. Nuciferine is able to alleviate hyperuricemia significantly. However, the underlying metabolic regulation mechanism remains unknown. To understand the metabolic effects of nuciferine on hyperuricemia by establishing a rat model of rapid hyperuricemia, 1H NMR and liquid chromatography-mass spectrometry were used to conduct nontargeted metabolomics studies. A total of 21 metabolites were authenticated in plasma and urine to be closely related with hyperuricemia, which were mainly correlated to the six metabolic pathways. Moreover, 16S rRNA analysis indicated that diversified intestinal microorganisms are closely related to changes in differential metabolites, especially bacteria from Firmicutes and Bacteroidetes. We propose that indoxyl sulfate and N-acetylglutamate in urine may be the potential biomarkers besides uric acid for early diagnosis and prevention of hyperuricemia. Gut microbiological analysis found that changes in the gut microbiota are closely related to these metabolites.

A database for risk assessment and comparative genomic analysis of foodborne Vibrio parahaemolyticus in China
Vibrio parahaemolyticus is a major foodborne pathogen worldwide. The increasing number of cases of V. parahaemolyticus infections in China indicates an urgent need to evaluate the prevalence and genetic diversity of this pathogenic bacterium. In this paper, we introduce the Foodborne Vibrio parahaemolyticus genome database (FVPGD), the first scientific database of foodborne V. parahaemolyticus distribution and genomic data in China, based on our previous investigations of V. parahaemolyticus contamination in different kinds of food samples across China from 2011 to 2016. The dataset includes records of 2,499 food samples and 643 V. parahaemolyticus strains from supermarkets and marketplaces distributed over 39 cities in China; 268 whole-genome sequences have been deposited in this database. A spatial view on the risk situations of V. parahaemolyticus contamination in different food types is provided. Additionally, the database provides a functional interface of sequence BLAST, core genome multilocus sequence typing, and phylogenetic analysis. The database will become a powerful tool for risk assessment and outbreak investigations of foodborne pathogens in China.

Different Exosomal microRNA Profile in Aquaporin-4 Antibody Positive Neuromyelitis Optica Spectrum Disorders
Neuromyelitis optica spectrum disorders (NMOSD) and multiple sclerosis (MS) are inflammatory demyelinating diseases of the central nervous system. Exosomal microRNAs (miRNAs) are emerging biomarkers for demyelinating diseases. In this study, 52 aquaporin-4 antibody serum-positive NMOSD patients, 18 relapsing-remitting multiple sclerosis (RRMS) patients and 17 healthy controls (HCs) were included for the next-generation sequencing (NGS). To validate the NGS results, the valuable miRNAs were selected for validation by real-time quantitative polymerase chain reaction in another cohort of patients, comprising 31 NMOSD patients and 14 HCs. In addition, these miRNAs were also validated in a longitudinal study. NGS data revealed the exosomal miRNAs profile in NMOSD patients was different from HCs. Among those potential exosomal miRNAs which can distinguish NMOSD status, hsa-miR-122-3p and hsa-miR-200a-5p were the most abundant miRNAs. In addition, hsa-miR-122-3p and hsa-miR-200a-5p were significantly upregulated in the serum exosome of relapsing NMOSD compared with that in remitting NMOSD. Hsa-miR-122-3p and hsa-miR-200a-5p had positive correlations with disease severity in NMOSD patients. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that the MAPK, Wnt and Ras signaling pathways were enriched. Further biological function analysis demonstrated that these two miRNAs might be involved in the immunoregulation of NMOSD pathogenesis. Our results indicated that miRNAs delivered by exosomes could be applied as potential biomarkers for NMOSD.

Myelin oligodendrocyte glycoprotein-associated disorders are associated with HLA subtypes in a Chinese paediatric-onset cohort
Objective: Myelin oligodendrocyte glycoprotein-associated disorders (MOGADs) are a rare new neurological autoimmune disease with unclear pathogenesis. Since a linkage of the disease to the human leucocyte antigen (HLA) has not been shown, we here investigated whether MOGAD is associated with the HLA locus.
Methods: HLA genotypes of 95 patients with MOGADs, assessed between 2016 and 2018 from three academic centres, were compared with 481 healthy Chinese Han individuals. Patients with MOGADs included 51 paediatric-onset and 44 adult-onset cases. All patients were seropositive for IgG targeting the myelin oligodendrocyte glycoprotein (MOG).
Results: Paediatric-onset MOGAD was associated with the DQB1*05:02–DRB1*16:02 alleles (OR=2.43; OR=3.28) or haplotype (OR=2.84) of HLA class II genes. The prevalence of these genotypes in patients with paediatric-onset MOGAD was significantly higher than healthy controls (padj=0.0154; padj=0.0221; padj=0.0331). By contrast, adult-onset MOGAD was not associated with any HLA genotype. Clinically, patients with the DQB1*05:02–DRB1*16:02 haplotype exhibited significantly higher expanded disability status scale scores at onset (p=0.004) and were more likely to undergo a disease relapse (p=0.030). HLA–peptide binding prediction algorithms and computational docking analysis provided supporting evidence for the close relationship between the MOG peptide subunit and DQB1*05:02 allele. In vitro results indicated that site-specific mutations of the predicted target sequence reduced the antigen–antibody binding, especially in the paediatric-onset group with DQB1*05:02 allele.
Conclusions: This study demonstrates a possible association between specific HLA class II alleles and paediatric-onset MOGAD, providing evidence for the conjecture that different aetiology and pathogenesis likely underlie paediatric-onset and adult-onset cases of MOGAD.

Whole-exome sequencing reveals the major genetic factors contributing to neuromyelitis optica spectrum disorder in Chinese patients with aquaporin 4-IgG seropositivity
Background and objective: Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune disease. Although genetic factors are involved in its pathogenesis, limited evidence is available in this area. The aim of the present study was to identify the major genetic factors contributing to NMOSD in Chinese patients with aquaporin 4 (AQP4)-IgG seropositivity.
Methods: Whole-exome sequencing (WES) was performed on 228 Chinese NMOSD patients seropositive for AQP4-IgG and 1400 healthy controls in Guangzhou, South China. Human leukocyte antigen (HLA) sequencing was also utilized. Genotype model and haplotype, gene burden, and enrichment analyses were conducted.
Results: A significant region of the HLA composition is on chromosome 6, and great variation was observed in DQB1, DQA2 and DQA1. HLA sequencing confirmed that the most significant allele was HLA-DQB1* 05:02 ( p < 0.01, odds ratio [OR] 3.73). The genotype model analysis revealed that HLA-DQB1* 05:02 was significantly associated with NMOSD in the additive effect model and dominant effect model (p < 0.05). The proportion of haplotype “HLA-DQB1* 05:02-DRB1* 15:01” was significantly greater in the NMOSD patients than the controls, at 8.42% and 1.23%, respectively (p < 0.001, OR 7.39). The gene burden analysis demonstrated that loss-of-function mutations in NOP16 were more common in the NMOSD patients (11.84%) than the controls (5.71%; p < 0.001, OR 2.22). The IgG1-G390R variant was significantly more common in NMOSD, and the rate of the T allele was 0.605 in patients and 0.345 in the controls (p < 0.01, OR 2.92). The enrichment analysis indicated that most of the genetic factors were mainly correlated with nervous and immune processes.
Conclusions: Human leukocyte antigen is highly correlated with NMOSD. NOP16 and IgG1-G390R play important roles in disease susceptibility.